1.Introdução
No contexto da bioinformática, muitas análises são conduzidas em linha de comando, dificultando assim a sua aplicação por pessoas sem o conhecimento adequado em computação. No intuito de possibilitar que outros usuários possam ter acesso ao processo de análise e expandi-las para outros estudos, desenvolvemos uma ferramenta em interface gráfica usando a ferramenta R Shiny.
Esta ferramenta encontra-se alocada em um servidor WEB (no domínio http://cetics.butantan.gov.br/shiny/ngs_shiny_app2/), e seu código encontra-se armazenado no repositório do GitLab (também alocado em http://cetics.butantan.gov.br). O código estatá disponível para download após a ferramenta ser publicada em revista científica.2.Ferramenta de análise
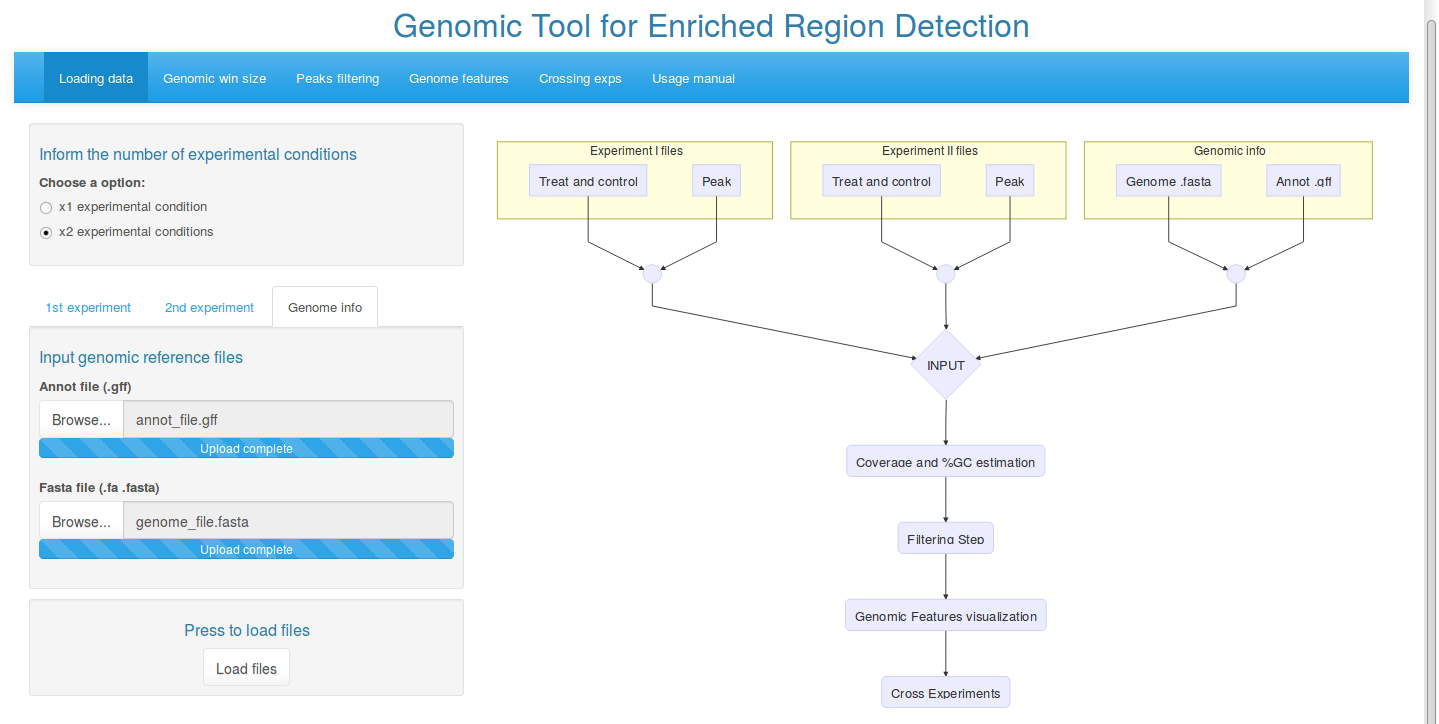
2.1.Upload dos dados
A ferramenta permite analisar até dois experimentos diferentes em pararelo. Os dados de entrada são compostos pelo arquivo de alinhamento das sequências (tratamento e controle em formato .bed, comprimidos em formato .zip), picos preditos pelo programa MACS2, genoma de referência (.fasta) e anotação genômica (.gff).
Exemplo de formato para cada tipo de entrada:
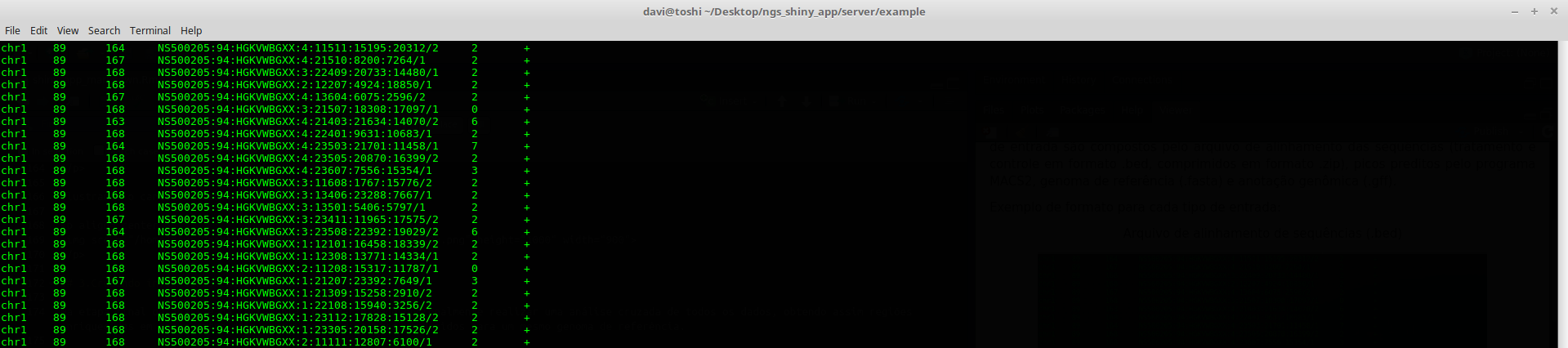
Arquivo de alinhamento de sequências (.bed)
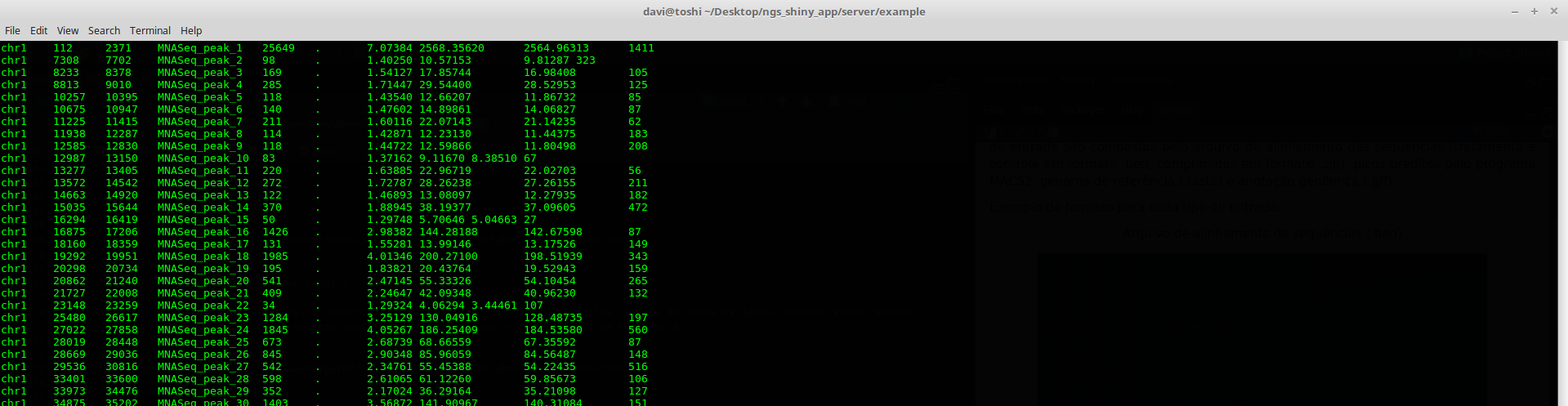
Arquivo dos picos pretidos pelo MACS2 (.narrowPeak)
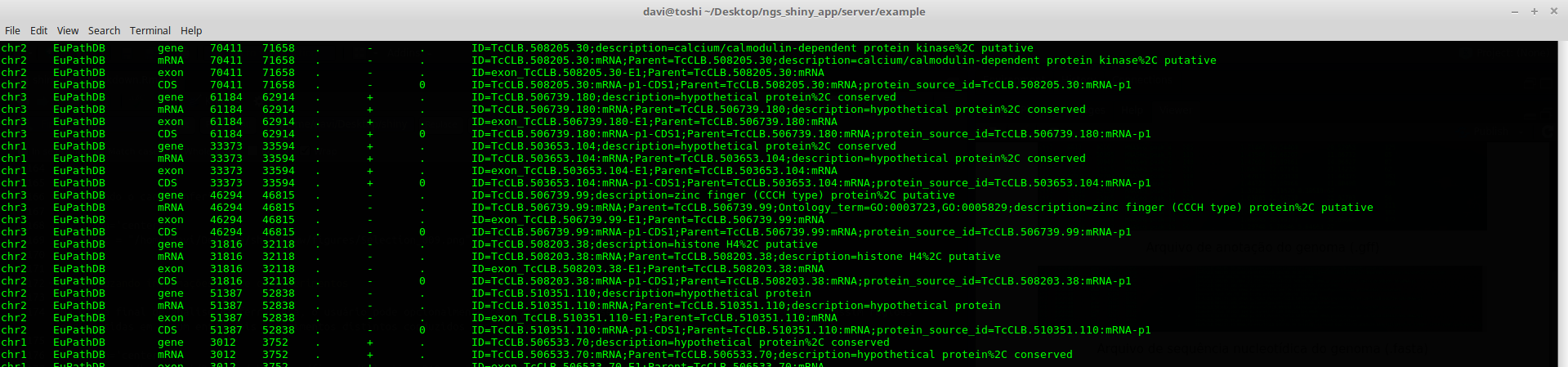
Arquivo de anotação do genoma (.gff)
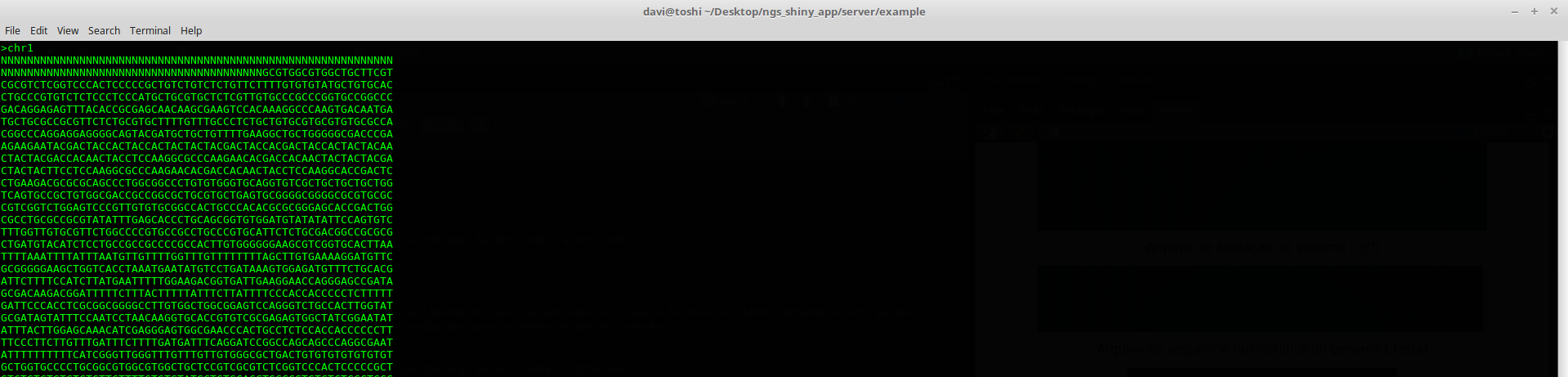
Arquivo de sequência nucleotídica do genoma (.fasta)
O usuário deve seguir cada etapa corretamente, afim de se obter os resultados da análise de regiões enriquecidas do genoma.
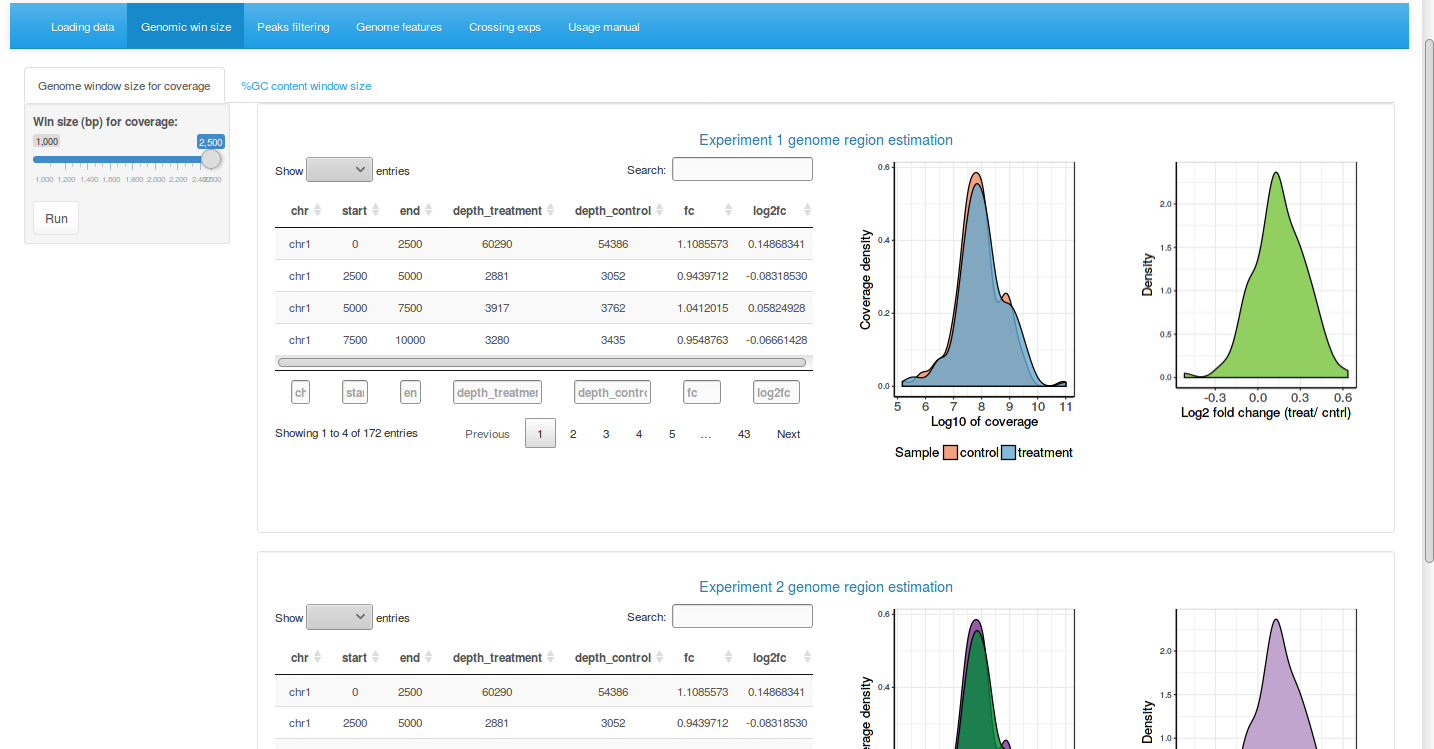
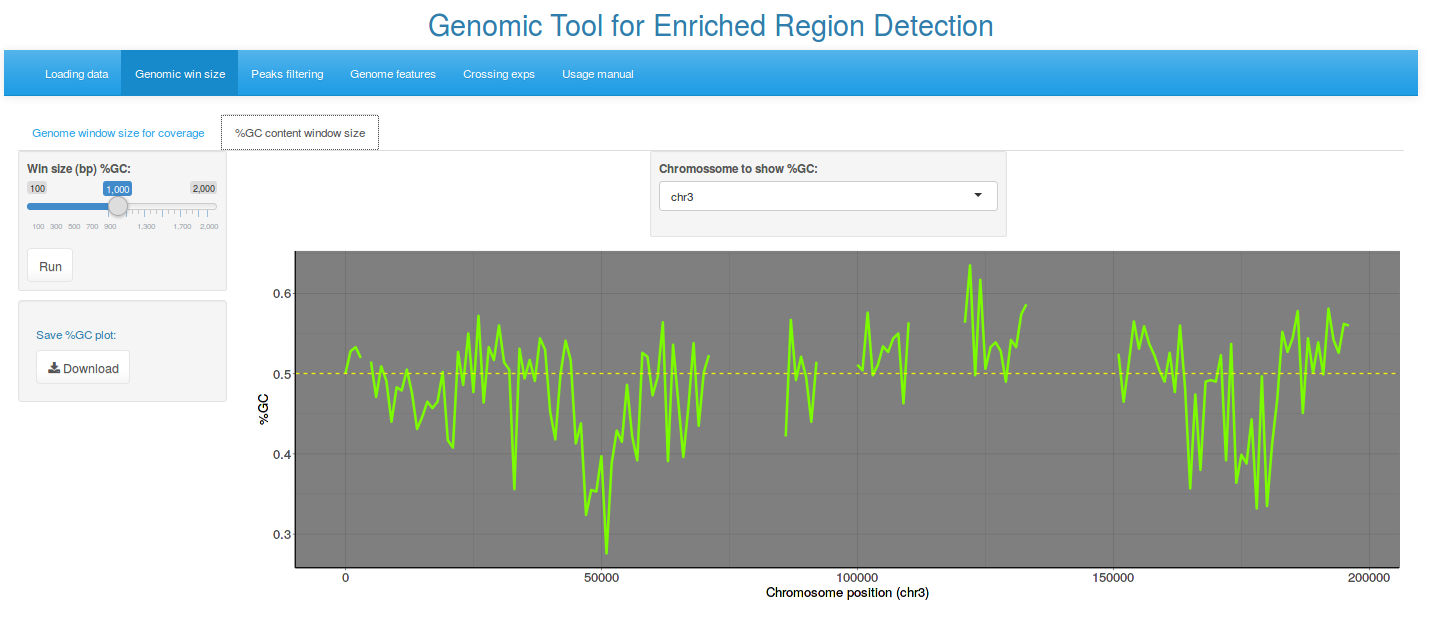
2.2. Estimando cobertura e conteúdo %GC
Após realizar o upload dos dados brutos, a ferramenta estima a cobertura genômica em janelas de tamanho definido pelo usuário, da mesma forma também é estimado o conteúdo GC.
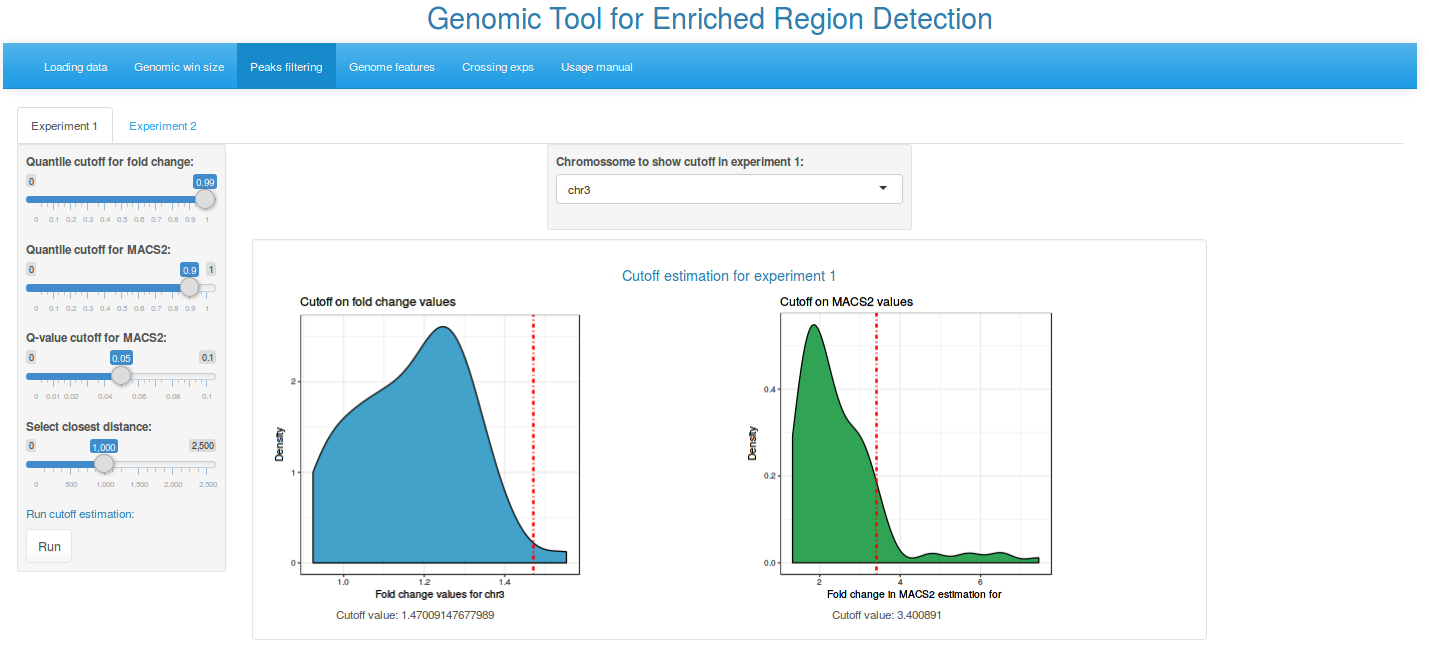
2.3. Aplicando filtro sobre os dados
Esta etapa é sucessidida pela aplicação de um filtro tanto sobre os dados de cobertura estimado, como também sobre dos dados dos picos preditos pelo programa MACS2.
2.4. Visualizando dados integrados
Tendo os dados filtrados em mãos, o usuário poderá gerar figuras de cada cromossomo de forma independente integrando as features de interesse tanto de um quanto de dois experimentos. Um menu dinâmico é criado conforme os dados experimentais.
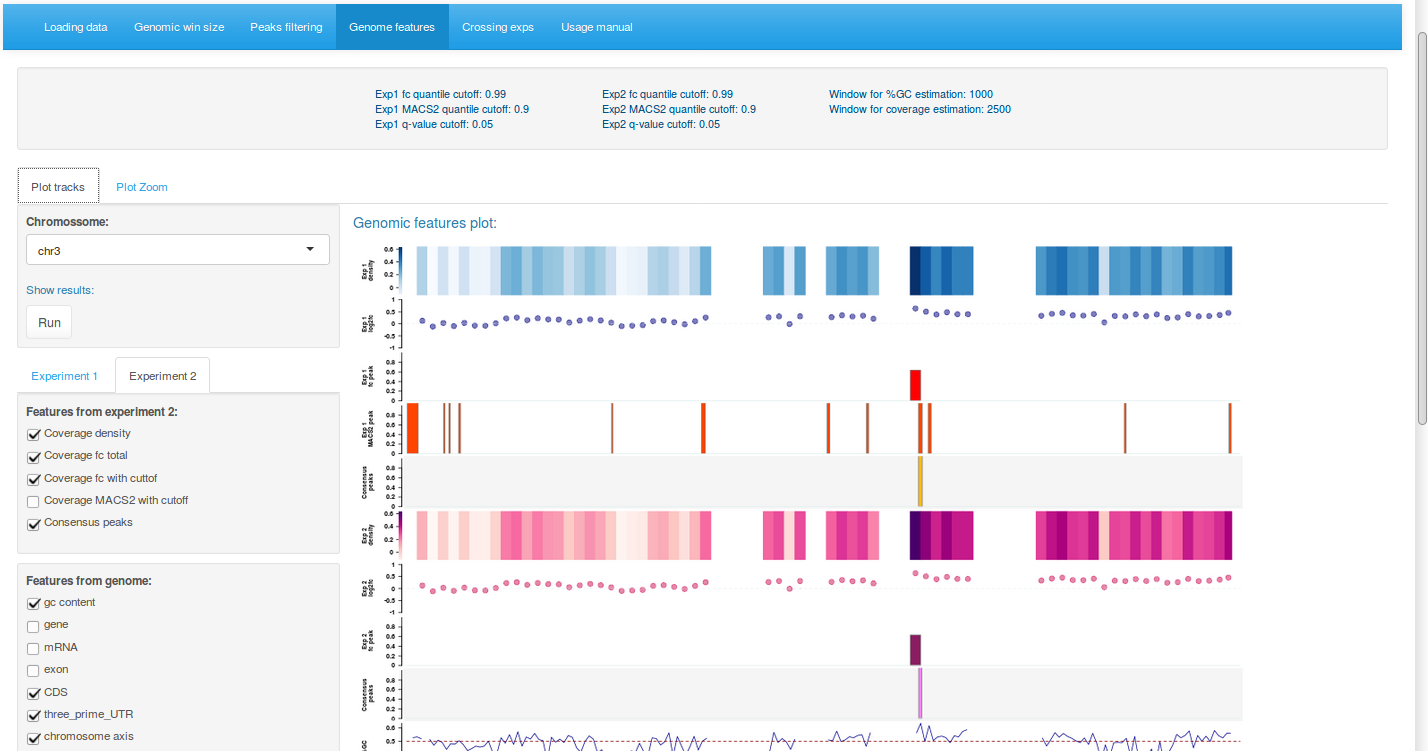
Ilustrando o canto inferior:
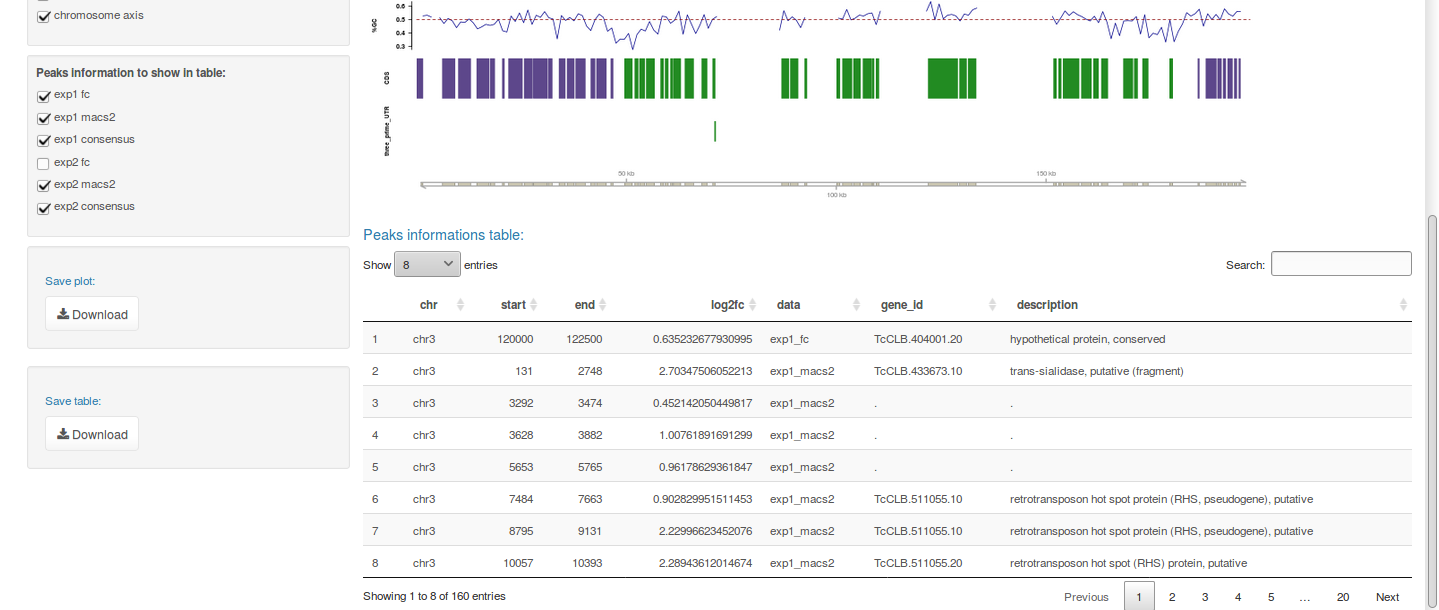
É possível ainda realizar um zoom sobre as regiões de interesse do cromossomo e destacar as características de interesse. Em todos os casos, é possível realizar o download tanto das figuras como também das informações contidas em uma tabela.
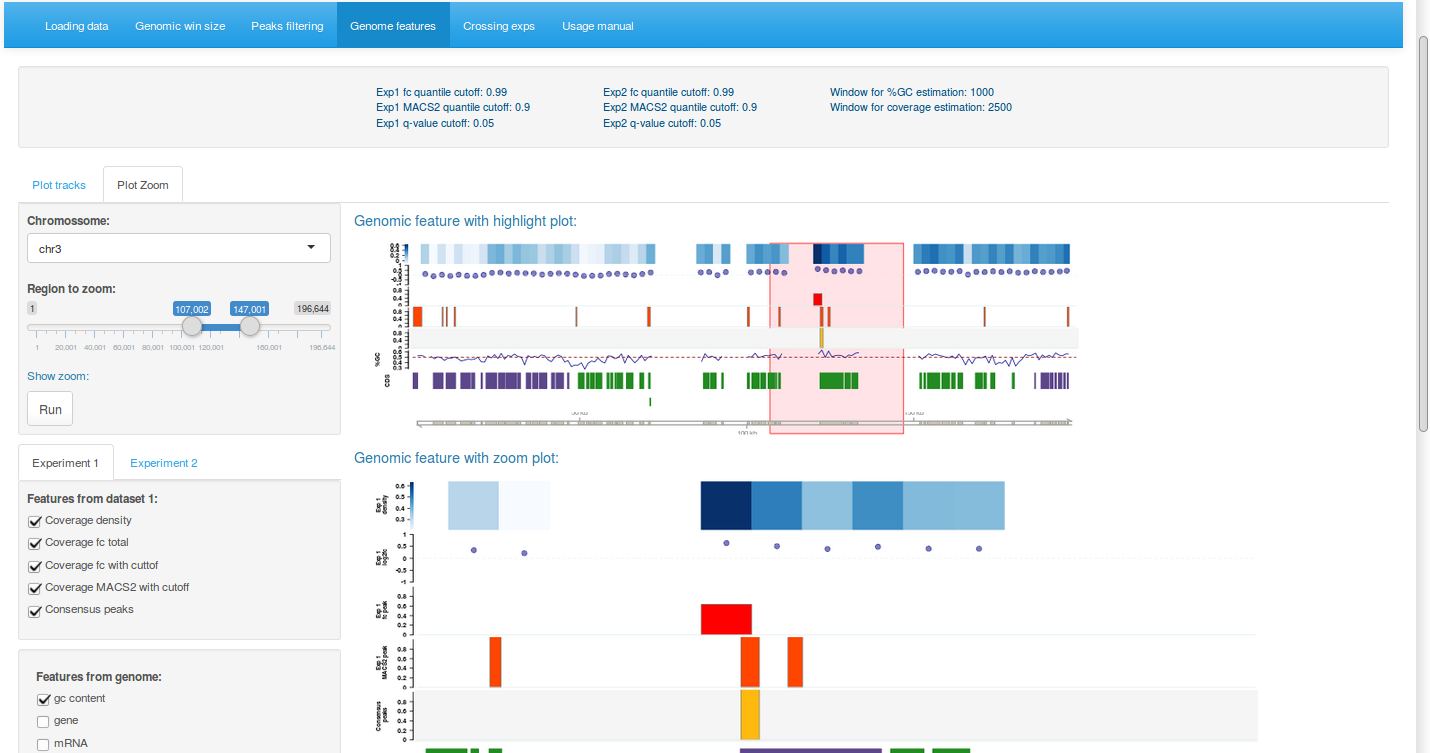
Ilustrando o canto inferior:
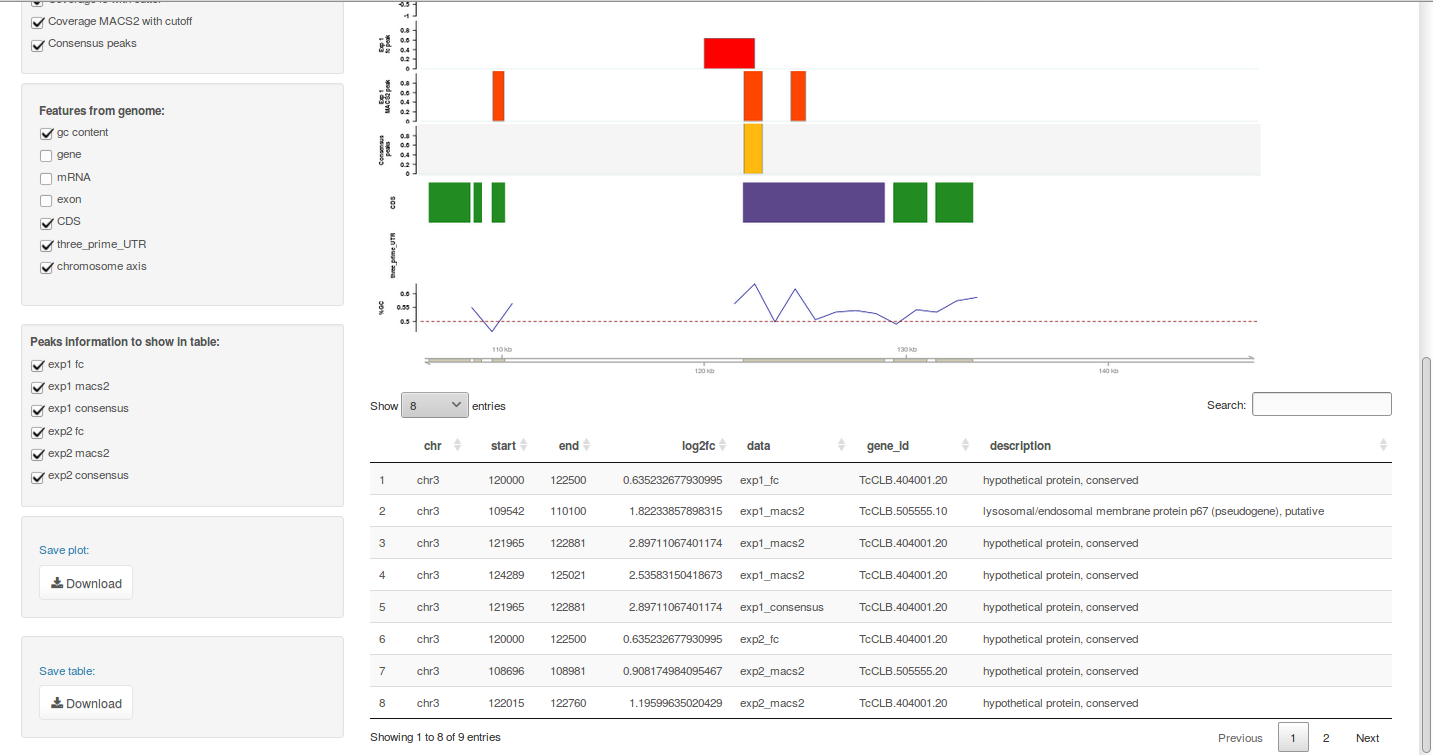
3.Cruzando informações entre os experimentos
Na etapa final de análise da ferramenta, o usuário pode opcionalmente realizar uma análise cruzada de todos os dados, obtendo assim regiões enriquecidas em comum entre dois experimentos distintos conduzidos para um mesmo genoma de referência.
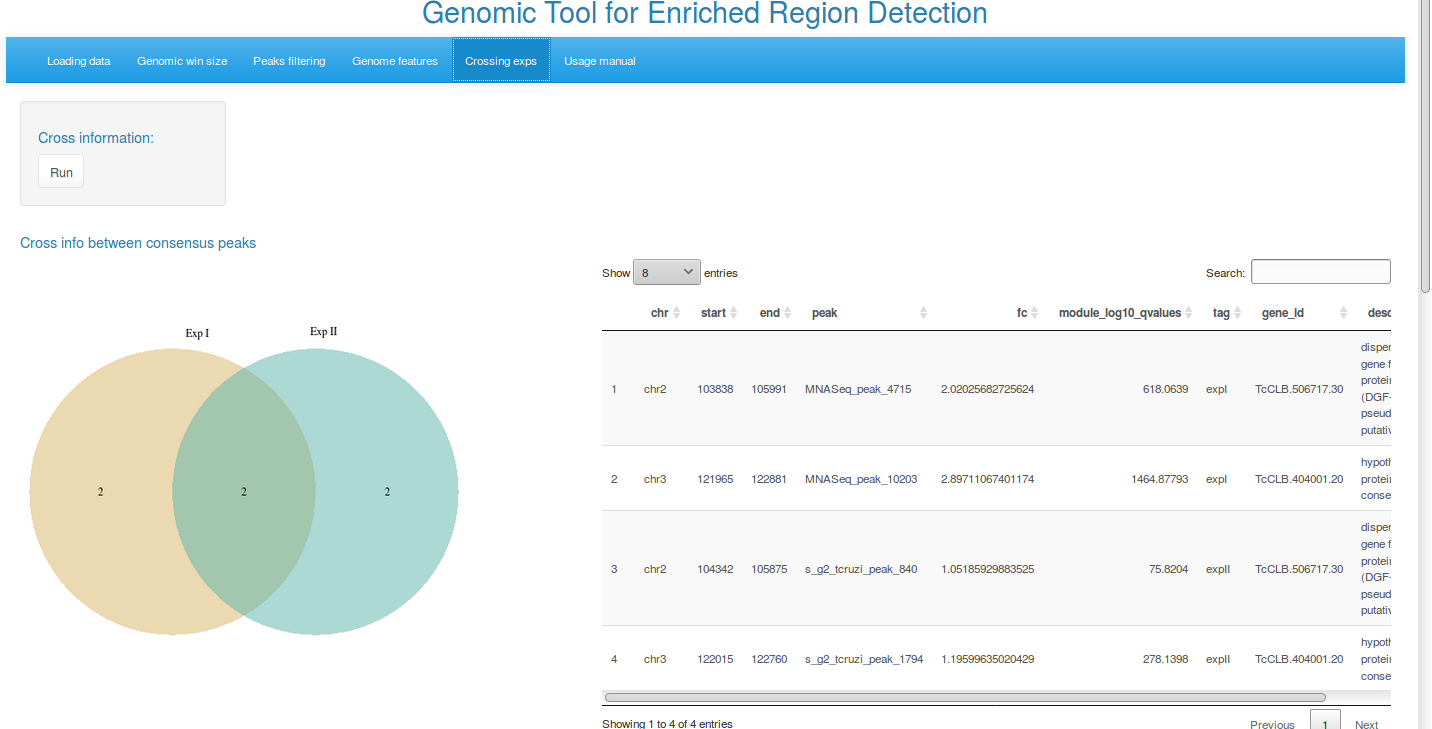
Ilustrando o canto inferior:
